経済産業省が2013年に「再生医療の実用化・産業化に関する研究会」で取りまとめた最終報告書に発表した再生医療の市場規模予測の図です。
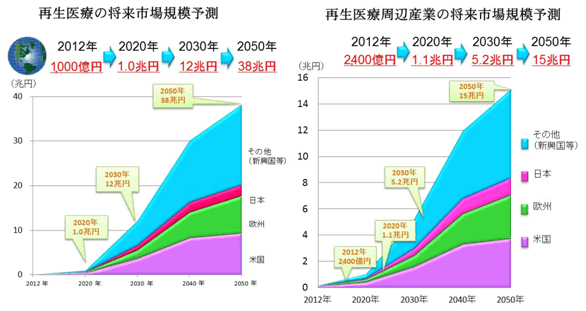
図1 出所) 経済産業省 NewsRelease 平成25年2月22日
この最終報告書において、2020年には、製造・加工品と周辺産業を合計すると、国内の再生医療の市場規模は1900億円、世界の再生医療の市場規模は2兆円と試算されています。
2012年の数値がそれぞれ280億円、3400億円であることを鑑みると6-7倍の伸びが予測されていました。
図1では、2012年、2020年、2030年、2050年の世界市場の予測値が記されています。
日本市場の数字を同じようにみると、再生医療の市場規模は、90億円→950億円→1.0兆円→2.5兆円、再生医療周辺産業については、170億円→950億円→5500兆円→1.3兆円と予測されています。
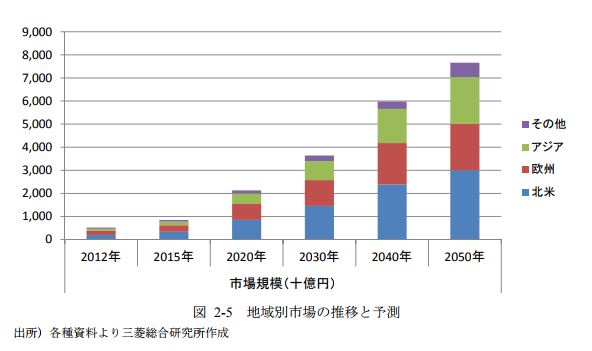
図2 出所) 株式会社三菱総合研究所 2015年3月31日 資料 *1)
図2は、市場の推移と予測です。
2020年以降の上昇率の違いはあれ、同じようなカーブを描いています。
上昇を支えるための策として、様々な提言がなされています。
前述の経済産業省の最終報告書でも、「今後の課題」として下記の6点が挙げられていました。
– 再生医療の治療の特徴に対する理解の促進
– 細胞加工機関に求められる基準とモデル契約書の作成
– 再生医療の審査手続きの合理化・透明化
– 再生医療の実用化のための技術開発
– 市場拡大に向けた業界団体の取り組みの活性化
– 再生医療の特性に適したリスク・費用負担の整備
また、国としても、省庁の枠を超えた取り組みを行おうとしています。
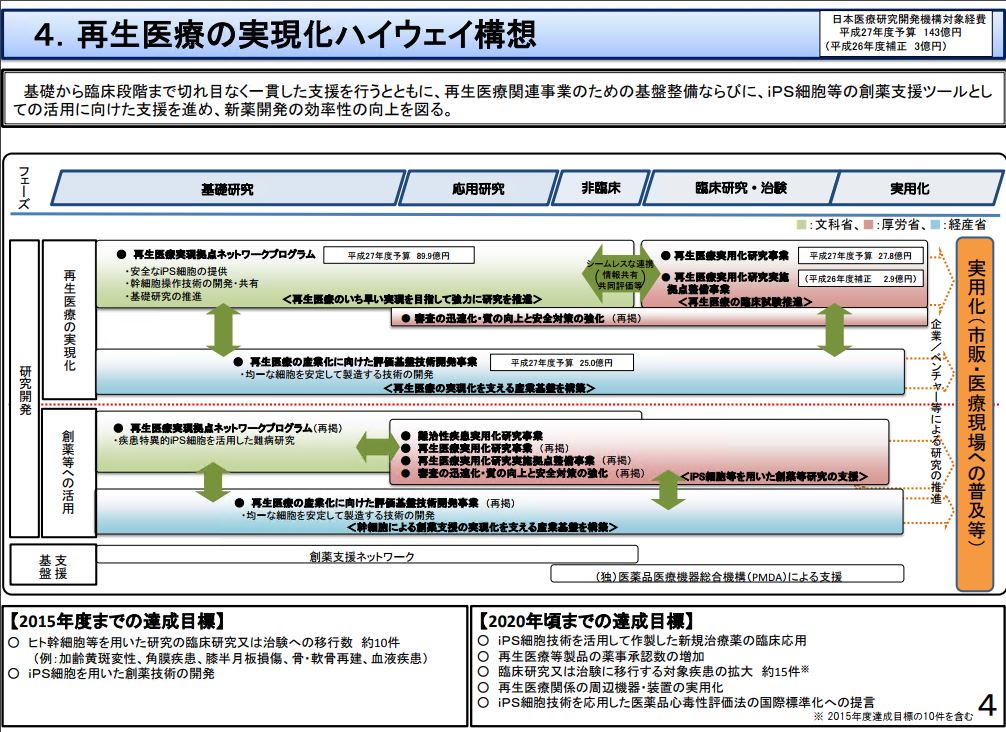
図3 出所) 平成27年度 医療分野の研究開発関連予算のポイント
倫理上の問題や手続きの整備等はもちろんですが、細胞ごとの培養法の確立と標準化、競争力ある消耗品(試薬、培地等)の開発と標準化が再生医療市場の拡大を後押しすることは確実だと思います。
「日本品質」を発揮することが、再生医療の市場規模と世界における日本のシェア拡大への王道ではないでしょうか。
世界基準を作る主導権を日本が勝ち取れたらいいですね。
*1)
株式会社三菱総合研究所 2015年3月31日 平成 26 年度「再生医療の産業化に向けた評価基盤技術開発事業(再生医療等の産業化に向けた評価手法等の開発)」(原料細胞の入手等に関する調査等)報告書